[LG]《Transformers Discover Molecular Structure Without Graph Priors》T Kreiman, Y Bai, F Atieh, E Weaver... [UC Berkeley] (2025)
图谱神经网络(GNN)长期主导分子机器学习,尤其在分子性质预测和机器学习势(MLIPs)中,但其依赖预定义图结构带来表达力限制和计算效率瓶颈。近期研究展示,未经修改的纯Transformer架构,直接以三维笛卡尔坐标为输入,且不依赖任何图结构或物理先验,也能实现分子能量与力的准确预测,且在同等计算预算下性能媲美甚至超越顶级GNN。
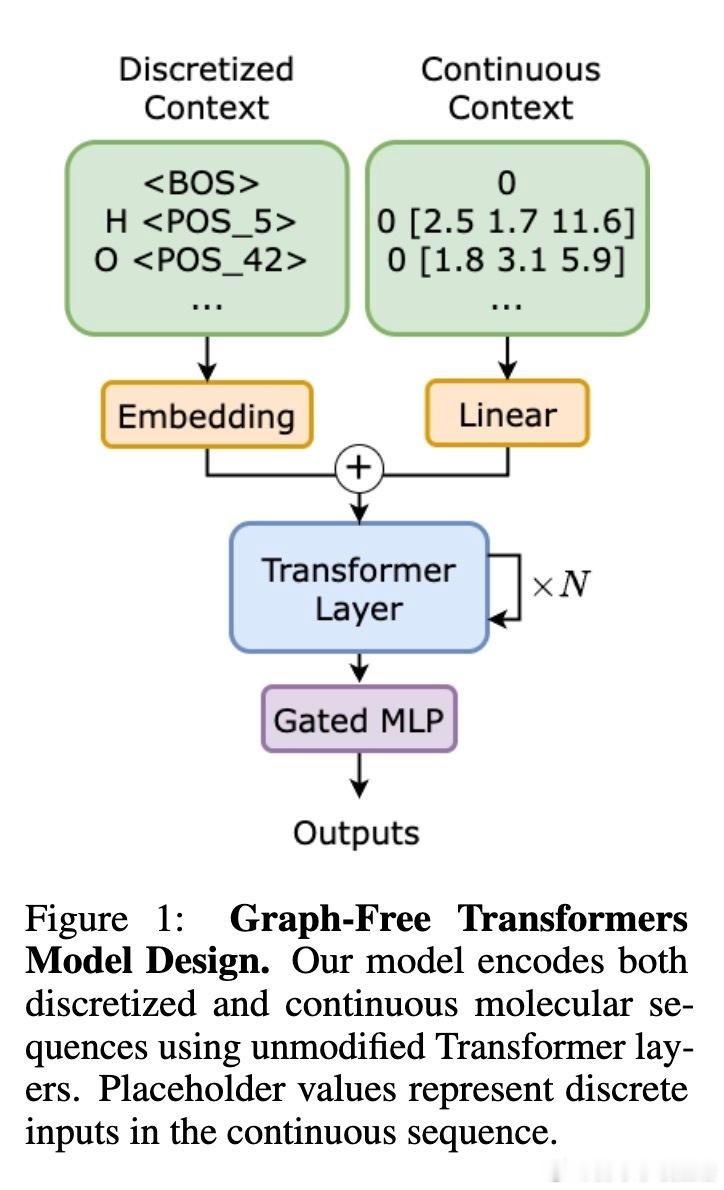
• 模型架构:基于LLaMA2标准Transformer,移除位置编码,融合离散和连续分子特征输入,保持原生多头自注意力机制不变,确保关系结构从数据中自适应学习,无硬编码偏置。
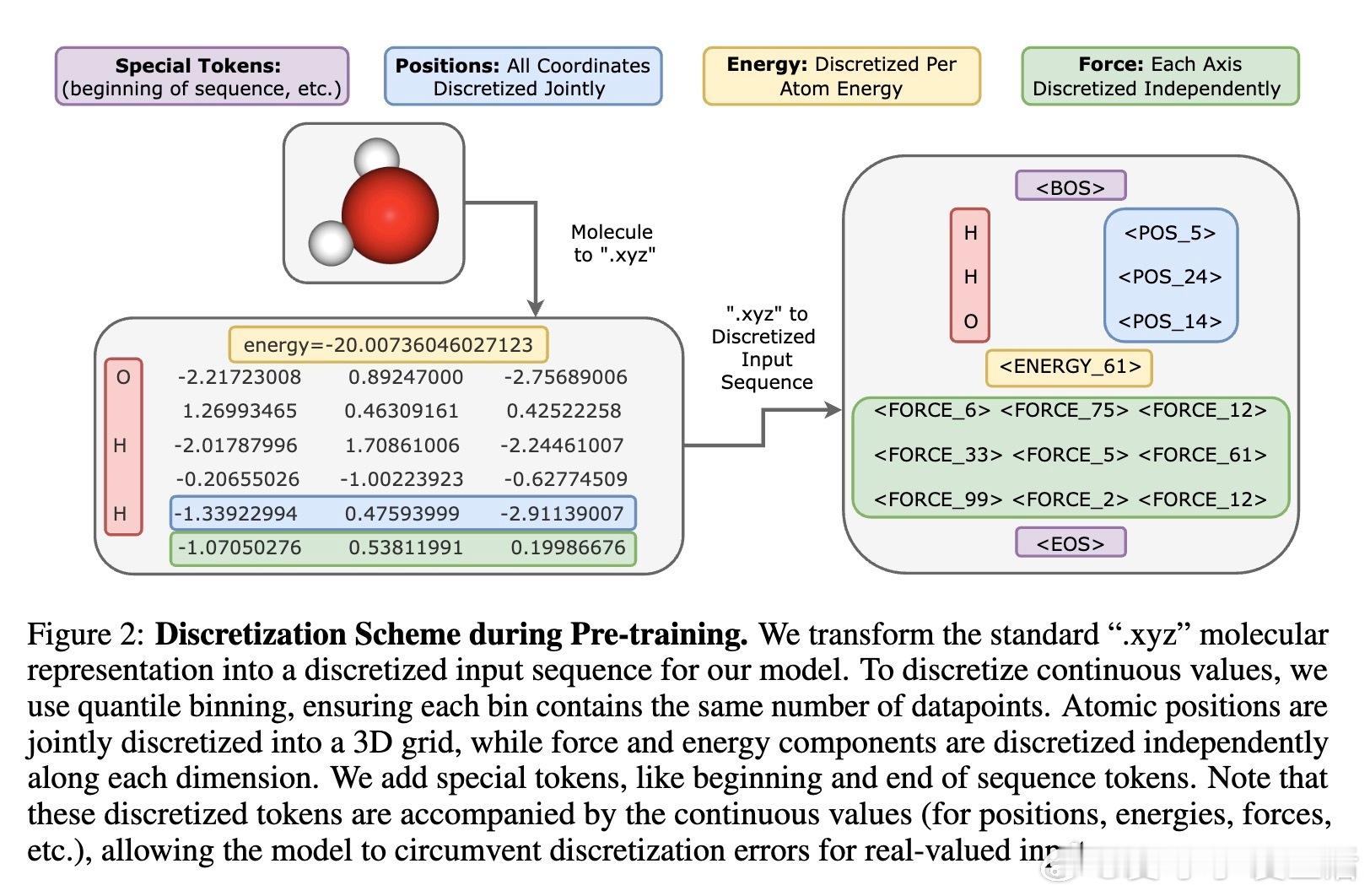
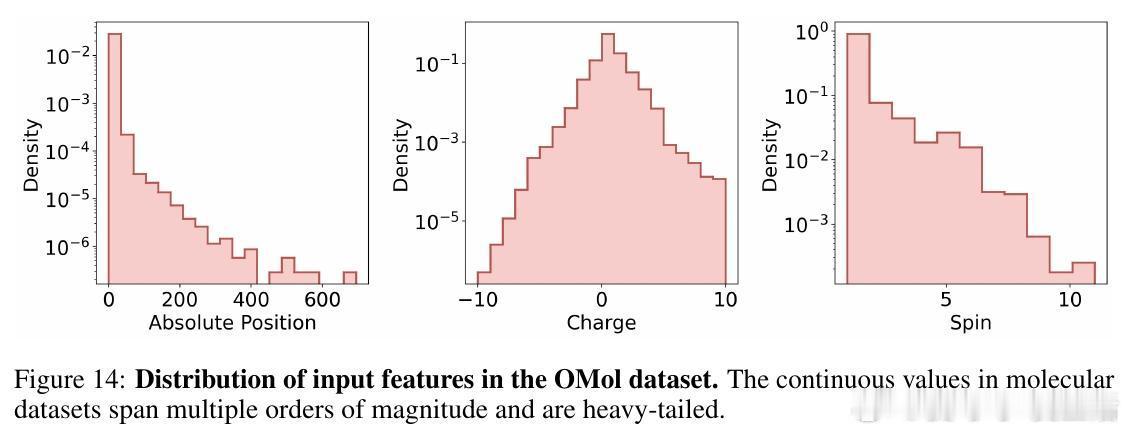
• 输入格式:采用分子.xyz格式,连续坐标等通过分位数离散化编码,同时附加连续数值减小离散误差,加入特殊标记控制预测任务(能量、力等)。
• 训练策略:两阶段——自回归预训练捕获位置、能量、力的联合分布(交叉熵损失),继以双向微调精确回归连续能量与力向量,实现置换等变性。
• 性能对比:在OMol25大规模数据集上,1B参数Transformer与6M参数的最先进等变GNN(eSEN)在能量和力预测均表现相近,且训练推理速度显著提升,得益于成熟的Transformer软件硬件生态。
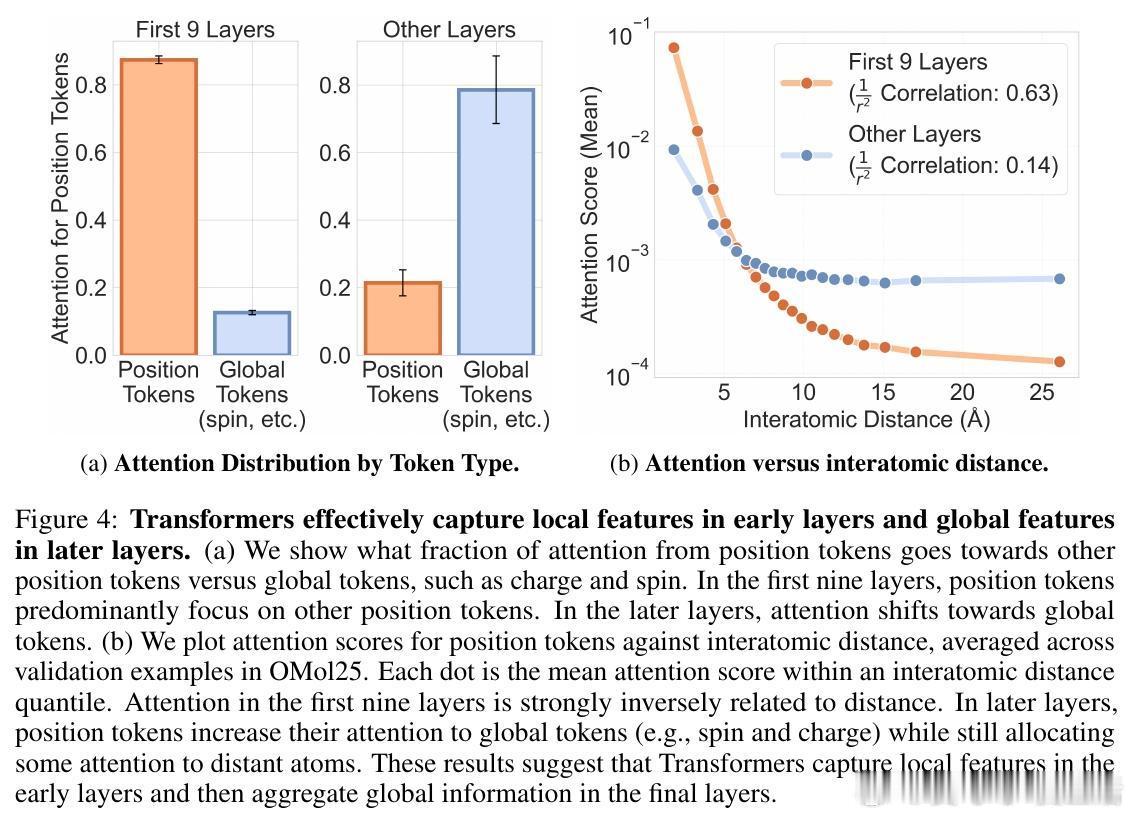
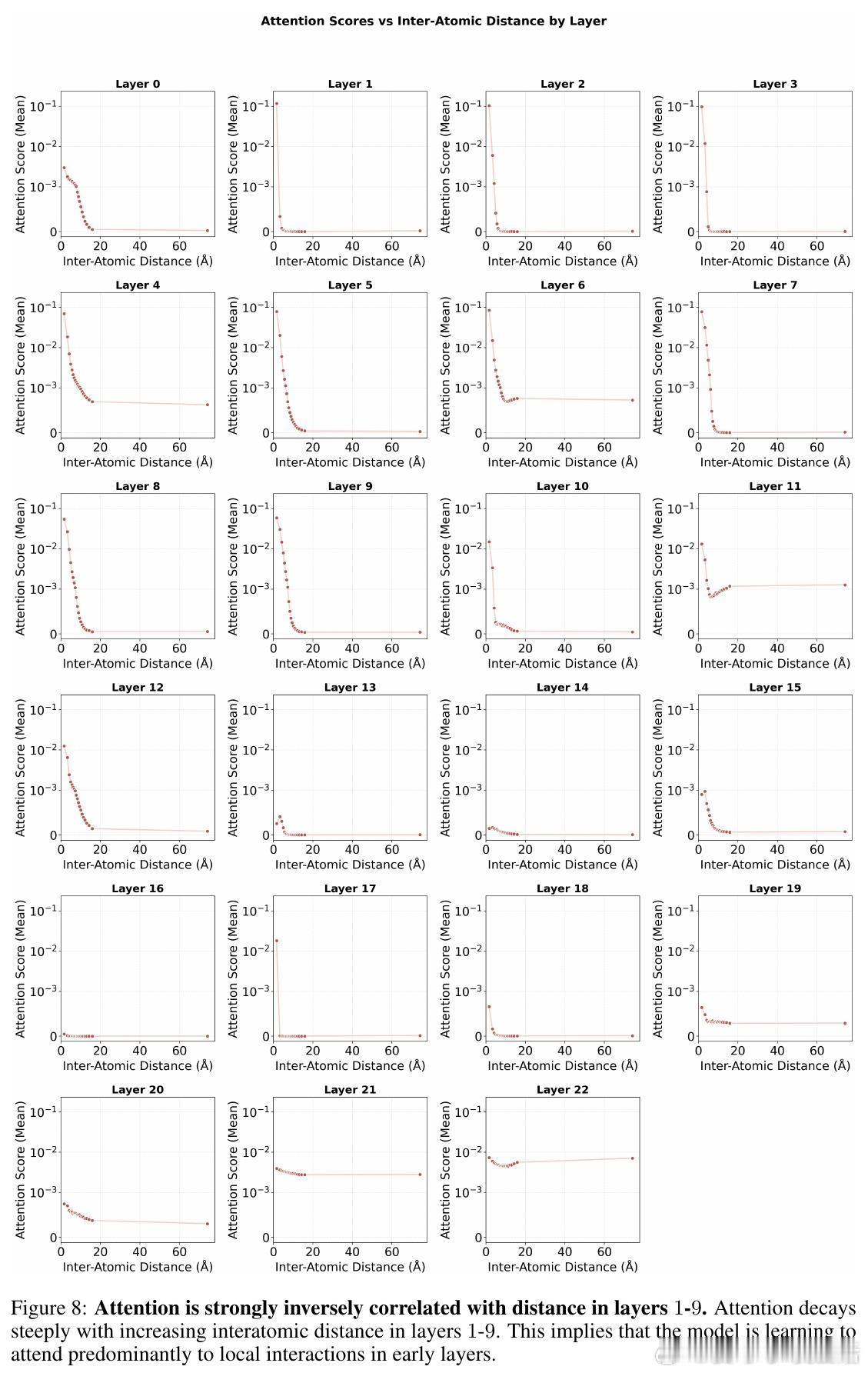
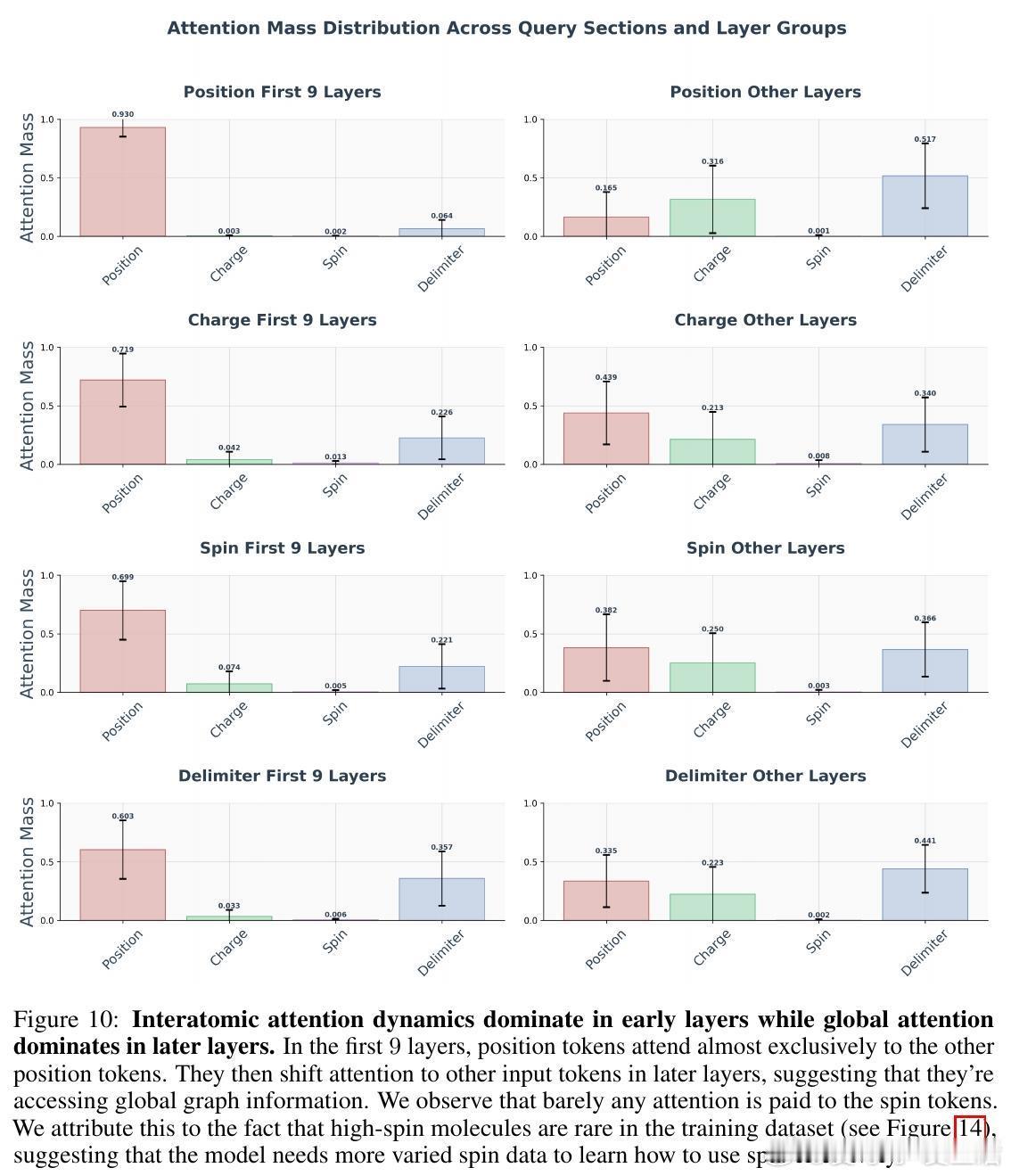
• 自适应注意力机制:Transformer早期层聚焦局部原子间距离,注意力强度与距离呈反比,后期层则转向全局分子属性(电荷、自旋),显示由局部到全局信息整合的层次性特征提取。
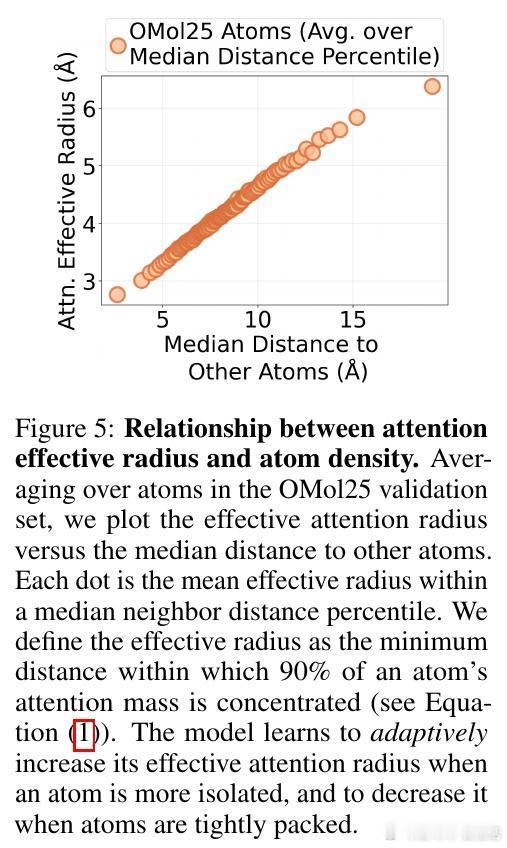
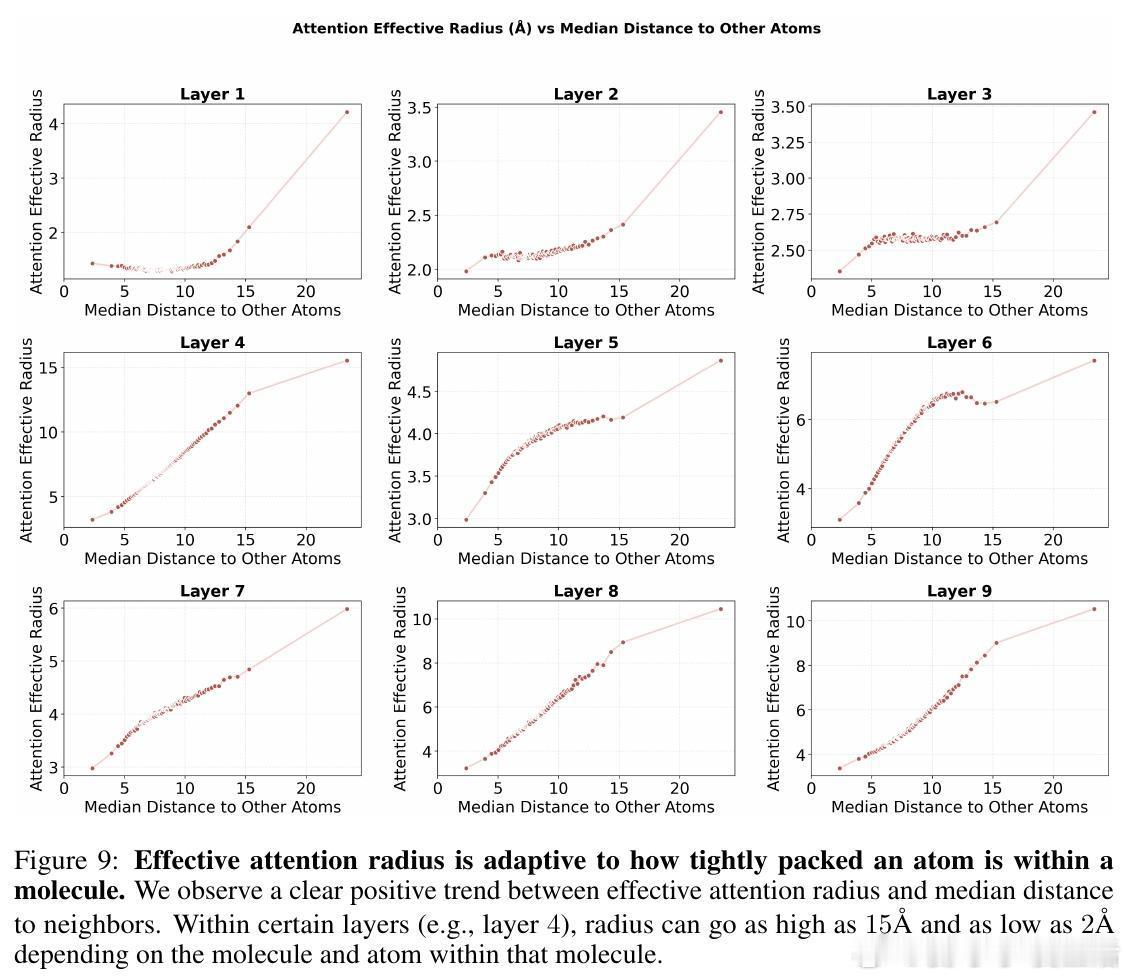
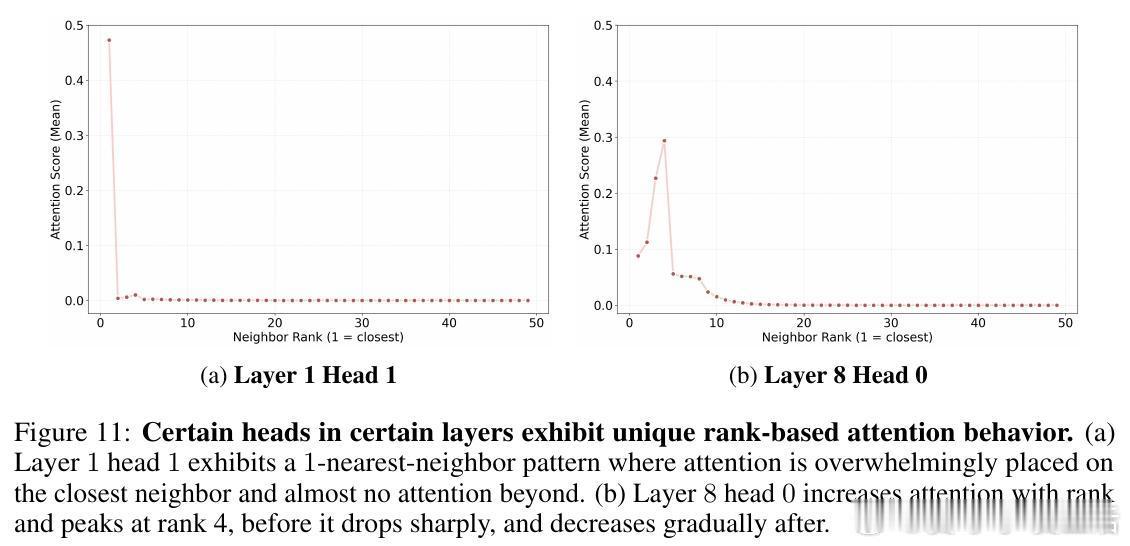
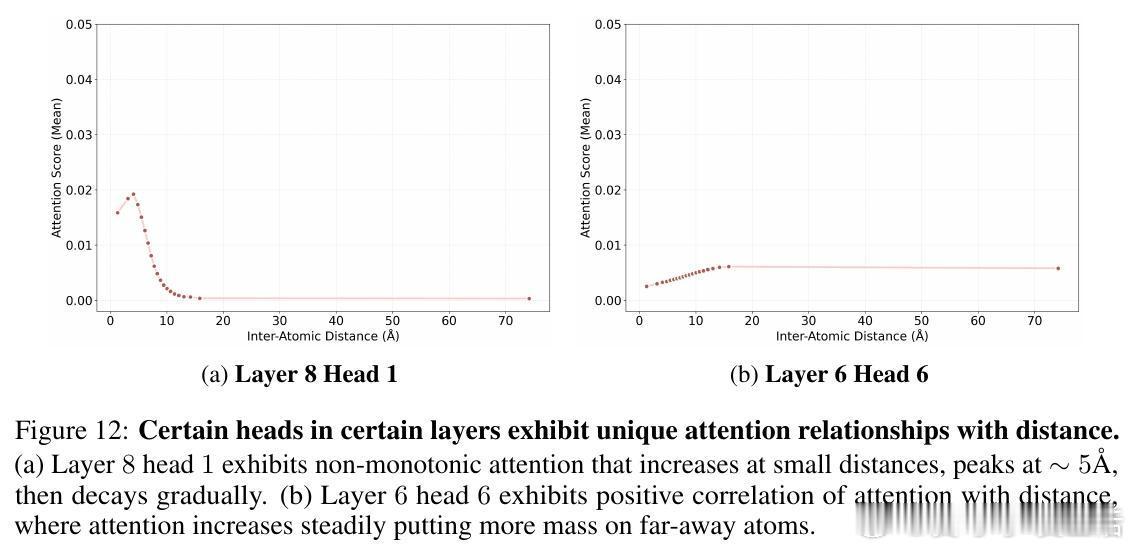
• 灵活的感受野:模型根据原子局部密度动态调整注意力有效半径,稀疏区域扩大关注范围,密集区域收缩,克服传统GNN固定半径限制,部分注意力头表现出k近邻或非单调距离依赖,体现复杂交互模式。
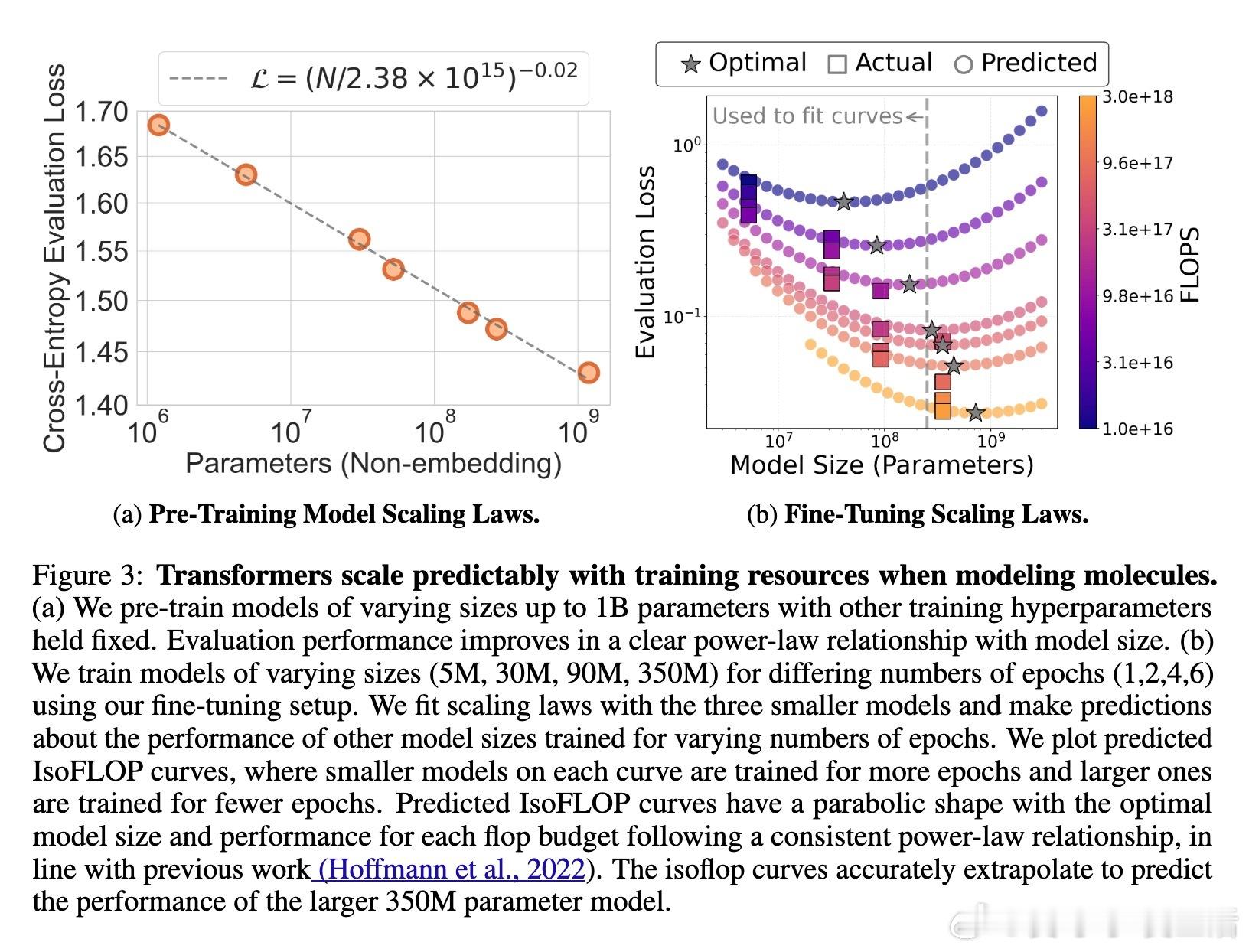
• 可扩展性与规律:模型性能遵循与语言和视觉领域类似的幂律扩展法则,规模提升带来持续性能改进,暗示未来可扩展至百亿参数级别。
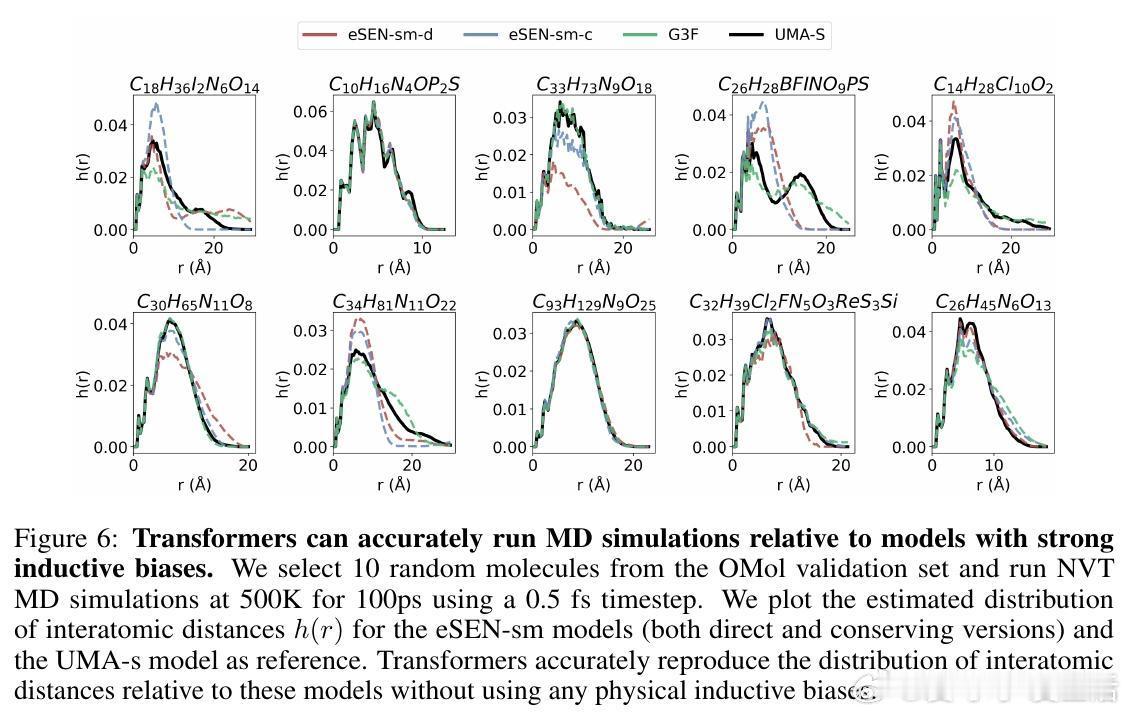
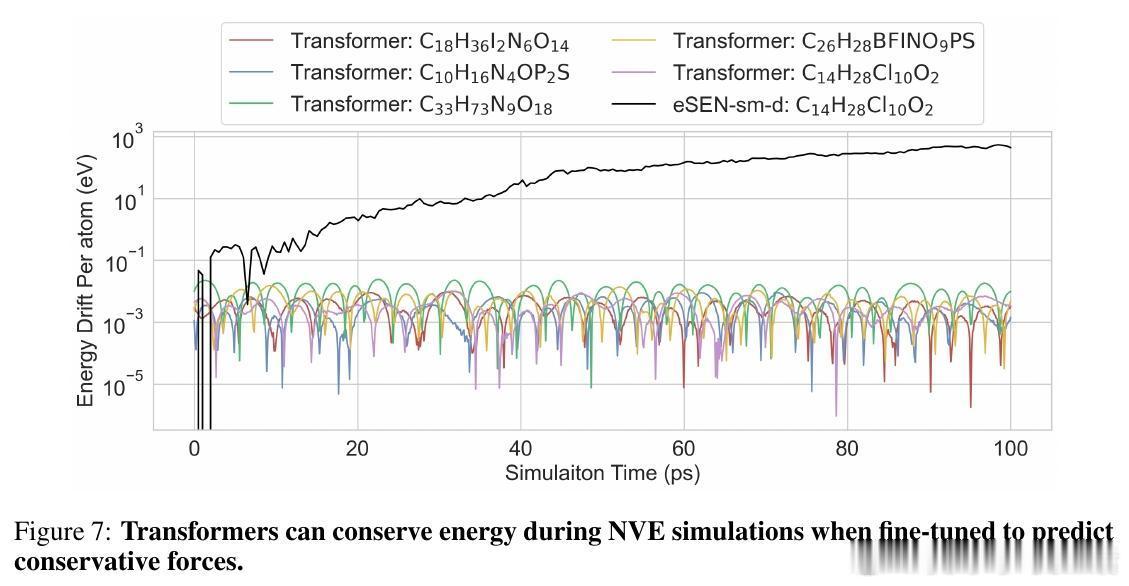
• 下游应用验证:Transformer成功执行分子动力学模拟,稳定运行NVT和能量守恒的NVE过程,具备实际物理模拟潜力。
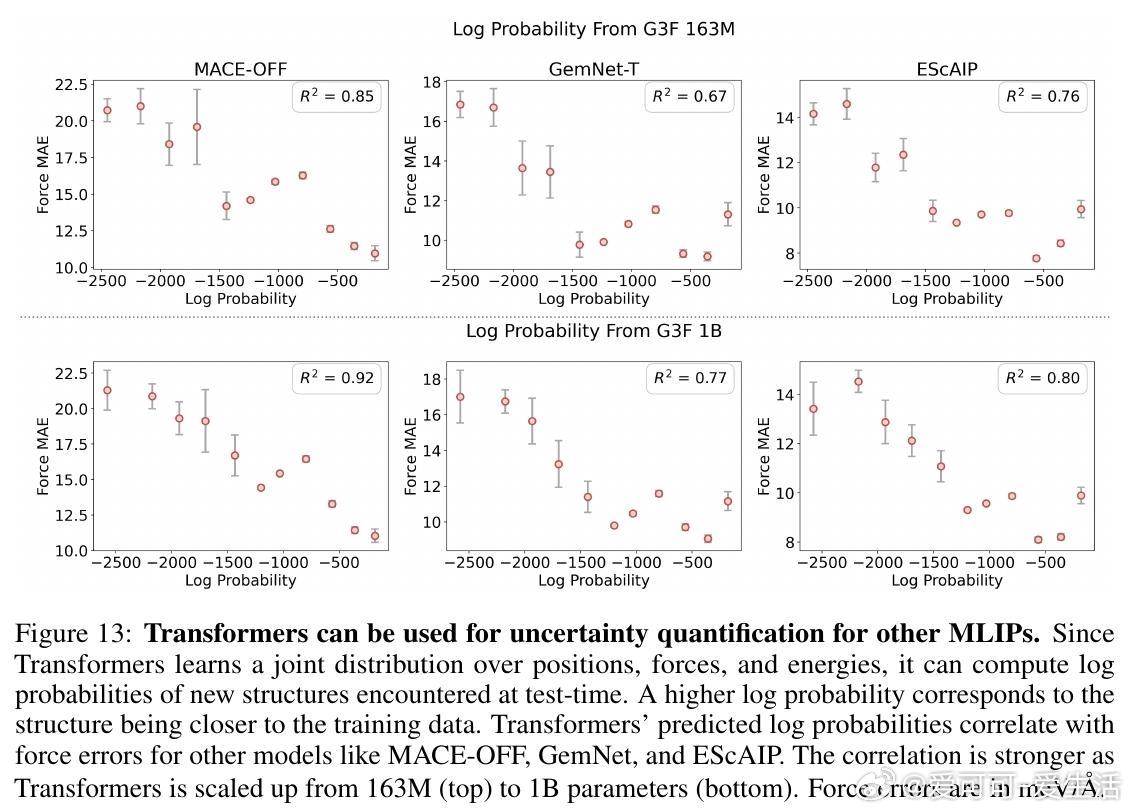
• 不确定性估计:通过学习联合分布,Transformer可输出结构的对数概率,用于识别训练数据外样本,辅助模型可信度评估。
心得:
1. 传统GNN的物理先验不再是必需,强大通用的Transformer可从数据中自发学习分子结构中的复杂物理规律与相互作用。
2. 自适应注意力机制突破了固定邻域限制,提供更灵活且更符合实际化学环境的感受野设计思路。
3. 统一的Transformer架构与成熟生态体系带来显著计算效率优势,为大规模分子建模和多模态集成奠定基础。
开创了无先验图结构的分子建模新范式,助推化学机器学习向更标准化、可扩展、灵活的方向发展。
🔗
详情🔗 tkreiman.github.io/projects/graph-free-transformers/
分子机器学习Transformer图神经网络机器学习势分子动力学计算化学